Legal & Safety Information (Byteflies Kit)
CE mark
Table of contents
LAST UPDATED: Jan 18, 2023
Introduction
The Byteflies Kit (Model 1) is a wearable CE Class I medical device for continuous recording of non-invasive physiological and behavioral signals in healthcare and home settings. The Byteflies Kit instructions for use (IFU) contains all the information required for safe and correct use of the Byteflies Kit. This page summarizes relevant safety and regulatory information.
- Byteflies Kit
- The collective name for all components of the medical device.
- Sensor Dot
- A 2-channel biopotential and tri-axial inertial measurement device, indicated for recording electroencephalography (EEG), electrocardiography (ECG), heart rate (HR), respiratory rate (RR), electromyography (EMG), electrooculography (EOG), acceleration (ACC), angular velocity (GYR), and activity index (AI).
- Docking Station
- A component that can charge up to 5 Sensor Dots and provide a secure communication gateway to the Byteflies Cloud to upload recorded data.
- Byteflies Cloud
- A secure data storage and processing system for data recorded by Sensor Dots.
Intended Use
The Byteflies Kit is a multisensor device intended to collect non-invasive physiological and behavioral signals to be used in the clinic and/or home for Users that need continuous monitoring.
Intended Users
- Trained Operator
- A healthcare professional trained in the usage of Byteflies Kit.
- User
- A patient or subject with a clinical need for biopotential and/or motion monitoring and/or their caregiver.
The Byteflies Kit is intended to be used by Trained Operators who configure the Byteflies Kit and its accessories in a medical environment (e.g. hospital). Users are wearers of the Sensor Dot, not operators of the Byteflies Kit, and are 3 years of age or older. Once the correct set up is configured, the User can take the Byteflies Kit home to continue recording data as instructed by the Trained Operator. The User may be required to return to the medical environment for removal or reconfiguration of Byteflies Kit components by the Trained Operator.
Clinical Indications
The Byteflies Kit is intended for prescription use in the home or healthcare facility to acquire, record, and transmit physiological and behavioral signals from Users. It acquires, records and transmits electroencephalogram (EEG), electrocardiogram (ECG), heart rate (HR), respiratory rate (RR), electromyogram (EMG), and/or electrooculogram (EOG) signals, with optional accelerometer (ACC), gyroscope (GYR), and activity index (AI) signals. It only acquires these signals, no diagnostic claims are made about the User’s condition.
![]() The Byteflies Kit should not be used on Users that:
The Byteflies Kit should not be used on Users that:
- Have been diagnosed with or are suspected to have life-threatening conditions that could result in immediate danger;
- Need to undergo an MRI scan or cardiac defibrillation;
- Have an active implanted device such as a pacemaker, defibrillator, or vagal nerve stimulator;
- Are between 0 and 3 years of age.
Safety Information
The following is important information for using the Byteflies Kit properly and safely. Carefully read this section before operating any component. The Byteflies Kit is a biopotential and motion recording device comprised of hardware and software that has been tested to assess its safety and effectiveness, and to establish substantial equivalence with predicate devices.
- The Byteflies Kit cannot provide diagnoses. It only records physiological and behavioral signals as configured by and under the authority of the Trained Operator.
- The Sensor Dot LED patterns only reflect battery status and operation mode. The LED does not reflect physiological status in any way and should never be interpreted as an indication of patient health.
- The Byteflies Kit is designed to be operated by a Trained Operator. The User should always receive proper instructions from a Trained Operator before using any component of the Byteflies Kit.
- All components of the Byteflies Kit are only intended to be used by the User as instructed by the Trained Operator and the User cannot configure any component of the Byteflies Kit.
- The data recorded by a Sensor Dot can only be used to asses heart, brain, muscle, or motion-related diseases when reviewed by a properly trained healthcare professional (e.g. a cardiologist, pulmonologist, neurologist, physiologist …).
- Sensor Dots interface with the human body via Cradles. Cradles for monitoring of biopotential (ExG) signals are compatible with certain commercial ExG electrodes, such as but not limited to Byteflies ExG Adhesives. These commercial electrodes are not supplied as part of the Byteflies Kit. The use of any skin adhesives may cause mild skin irritation, such as redness, itching, or contact dermatitis in some Users. Consult the manufacturer of the ExG electrodes for more information.
Warnings

Do not use any component of the Byteflies Kit before reading these warnings, the IFU and -if applicable- a manual for the ExG electrodes:
- Do not use the Byteflies Kit if any component is damaged.
- The Byteflies Kit is provided non-sterile.
- Please verify the LED status indicators to avoid misuse.
- If the Trained Operator or User experience any adverse events while using a Byteflies Kit, discontinue the use and contact Byteflies immediately.
- Skin adhesives should be replaced if they no longer stick firmly to the skin or as indicated in their respective manuals.
- Similar devices may cause signal interference during data transmission. Avoid operating the Byteflies Kit near interfering devices.
- Keep the Byteflies Kit away from children and pets. The Sensor Dot and other small components of the Byteflies Kit may present a choking hazard, never place or let anyone place a component of the device in their mouth.
- If any component of the Byteflies Kit fails to operate after attempting all suggested troubleshooting steps, contact support as soon as possible.
- The battery used in a Sensor Dot may present a risk of fire, explosion, or chemical burn if mistreated. Do not expose Sensor Dots to excessive heat or fire. Do not crush, puncture, or incinerate as doing so can result in fire, explosion or the release of toxic gasses. Do not use or charge if a Sensor Dot appears to be leaking, discolored, deformed, or in any way abnormal.
- All components of a Byteflies Kit should be returned to the healthcare provider or study staff at the conclusion of the prescribed period of use.
- Do not use a Sensor Dot on a User without first cleaning it according to the instructions.
- Do not use a Byteflies Kit without first charging the Sensor Dot(s) and completing proper set up as described in the IFU.
- Do not use a Sensor Dot on Users undergoing an MRI scan.
- Do not use a Sensor Dot on Users undergoing defibrillation.
- Do not use a Sensor Dot on Users with an active implanted device, such as a pacemaker, defibrillator, or vagal nerve stimulator.
- Do not expose a Sensor Dot to strong sources of static electricity.
- Do not expose the Byteflies Kit to strong electromagnetic fields. They can distort the measurement. A minimal distance of 30cm should be kept from such fields.
- Do not leave a Sensor Dot on top of or next to other electrical equipment.
- To ensure a stable Byteflies Cloud connection, it’s important to install the Docking Station in an area with good signal strength. Keep in mind that obstacles like building structures can weaken the connection.
- To ensure uninterrupted connectivity and reliable transfer of recordings to the Byteflies Cloud, it is important to avoid installing the Docking Station near devices that emit radio waves like microwaves, areas with many obstacles, or places with weak Wi-Fi signal strength. Failure to do so may result in frequent connection drops and delayed recordings. However, in such cases, the recordings are saved on the Docking Station and uploaded to the Byteflies Cloud once a stable connection is established.
- The Docking Station Wi-Fi radio isn’t compatible with 5GHz Wi-Fi networks.
- Relocate or stop using the Byteflies Kit if it causes radio wave interference to a different wireless station.
- Do not submerge any component of a Byteflies Kit in liquid.
- Do not clean a Sensor Dot with agents other than those listed in the cleaning instructions.
- Do not damage any component of the Byteflies Kit through drops, violent shaking or crushing.
- Do not allow Sensor Dots to come into contact with conductive materials, except when following instructions on how to recharge the battery.
- Sensor Dot is not a toy. Usage on children 3 years of age and older should be under strict supervision of an adult.
- Do not alter any component of the Byteflies Kit. Any modification is strictly prohibited.
- Do not use at temperatures lower than 15°C or greater than 35°C.
- Do not use where air humidity is lower than 10% or higher than 90%.
- Do not use in environments subjected to pressure lower than 700 hPa or greater than 1080 hPa.
- Do not use a Docking Station with a charger and charging cable other than the one supplied with the Byteflies Kit.
Package and Device Symbols
| Symbol | Description |
|---|---|
 | Manufacturer |
 | Date of manufacture |
 | Catalog number |
 | Serial number |
 | Medical device |
 | Unique device identifier |
 | ID |
 | Version number |
 | CE marking of conformity |
 | Waste Electrical and Electronic Equipment (WEEE) compliance |
| Warning |
 | User should consult the instructions for use. |
 | Type BF Equipment (IEC 60601-1). Type BF is suitable for intentional external and internal application to the patient, excluding direct cardiac application. Type BF equipment has an F-type applied part. The applied part of the Byteflies Kit is the Sensor Dot component. |
 | For prescription use only |
 | Class II insulation |
 | Keep the device dry; the presence of liquids may compromise the safety of the device. |
 | The maximum and minimum temperature limits to which the device or accessory should be exposed during use. |
 | The maximum and minimum humidity limits to which the device or accessory should be exposed during use. |
 | The maximum and minimum atmospheric pressure limits to which the device or accessory should be exposed during use. |
 | Protected against solid objects over 12.5 mm, and protected against dripping water when tilted up to 15 degrees from its normal position. |
 | Includes RF transmitter; may cause RF interference. |
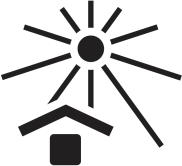 | Keep out of direct sunlight. |
 | The device is MRI unsafe; do not use the device in a magnetic resonance (MR) environment. |
 | Choking hazard for children ages 0-3 years. |
Regulatory Information
Declaration of Conformity
The Byteflies Kit (Model 1) is in conformity with the general safety and performance requirements of the EU Medical Device Regulation (MDR) 2017/45.
Byteflies Kit Compliant Regulatory Standards
- C
- The Byteflies Kit is compliant with this standard.
- R
- The Byteflies Kit references this standard ("in the spirit of").
| Standard | Implementation | Title | Remarks |
|---|---|---|---|
| 2017/745 EU MDR (2017) | C | EU Medical Device Regulation | |
| 2014/53/EU (2014) | C | Radio Equipment Directive | |
| EN ISO 13485:2016 + A11:2021 | C | Medical Devices - Quality Management Systems - Requirements For Regulatory Purposes | |
| FDA 21 CFR, part 820 | C | Quality system regulation for medical device, federal register | |
| EN ISO 14971:2019 + A11:2021 | C | Application of risk management to medical devices | |
| IEC 60601-1:2005 + AMD1:2012 + AMD2:2020 | C | Medical electrical equipment - Part 1: General requirements for safety | |
| IEC 60601-1-2:2014 (Ed. 4.0) + A1:2021 | C | Medical electrical equipment - part 1-2: General requirements for basic safety and essential performance - collateral standard: electromagnetic compatibility - requirements and tests | |
| IEC 60601-1-11:2015 + A1:2021 | C | Medical electrical equipment - Part 1-11: General requirements for basic safety and essential performance - Collateral Standard: Requirements for medical electrical equipment and medical electrical systems used in the home healthcare environment | |
| IEC 60601-1-6:2010 + A1:2013 | C | Medical electrical equipment - Part 1-6: General requirements for basic safety and essential performance - Collateral standard: Usability | |
| IEC 80601-2-26:2019 | C | Medical electrical equipment - Part 2-26: Particular requirements for the basic safety and essential performance of electroencephalographs | |
| IEC 60529:1989+ AMD1:1999+ AMD2:2013 | C | Degrees of protection provided by enclosures (IP code) | |
| IEC 62366-1:2015 + AMD1:2020 | C | Medical devices - Part 1: Application of usability engineering to medical devices | |
| IEC 62304:2006 + AMD1:2015 | C | Medical device software - Software life cycle processes + Amendment 1 | |
| ISO 20417:2021 | C | Medical devices - Information to be supplied by the manufacturer | |
| Regulation (EU) 2021/2226 on electronic instructions for use of Medical Devices (eIFU) | C | Electronic instructions for use of Medical Devices (eIFU) | |
| EN 62133-2:2017 + A1:2021 | C | Secondary cells and batteries containing alkaline or other non-acid electrolytes - Safety requirements for portable sealed secondary lithium cells, and for batteries made from them, for use in portable applications - Part 2: Lithium systems + Amendment 1 | |
| EN ISO 15223-1:2021 | C | Medical devices - Symbols to be used with medical device labels, labelling and information to be supplied - Part 1: General requirements | |
| EN ISO 14155:2011 | C | Clinical investigation of medical devices for human subjects – Good clinical practice | |
| ISO 10993-1:2018 | C | Biological evaluation of medical devices - Part 1: Evaluation and testing within a risk management process | |
| ISO 10993-5:2009 | C | Biological evaluation of medical devices - Part 5: Test for in vitro cytotoxicity | |
| ISO 10993-10:2010 | C | Biological evaluation of medical devices - Part 10: Tests for irritation and skin sensitization | |
| MEDDEV 2.7/1 Rev. 4 (2016) | C | Clinical Evaluation: Guide for manufacturers and notified bodies | |
| MEDDEV 2.12/1 rev 8 (2013) | C | Medical Devices Vigilance System | |
| MEDDEV 2.12/2 rev 2 (2012) | C | Clinical Evaluation - Post Market Clinical Follow-up | |
| MEDDEV 2.7/4 rev 12 (2010) | C | Guidelines on Clinical Investigation: A guide for manufacturers and notified bodies | |
| MEDDEV 2.7/3 rev 5 (2015) | C | Clinical investigations: serious adverse event reporting | |
| MDCG 202-5 rev 1 (April 2020) | C | Clinical Evaluation - Equivalence A guide for manufacturers and notified bodies | |
| MDCG 2019-11 | C | Qualification and classification of software - Regulation (EU) 2017/745 and Regulation (EU) 2017/746 | |
| MDCG 2019-15/rev.1 | C | Guidance notes for manufacturers of class I medical devices | |
| MDCG 2020-1 | C | Guidance on clinical evaluation (MDR) / Performance evaluation (IVDR) of medical device software | |
| MDCG 2020-7 | C | Post-market clinical follow-up (PMCF) Plan Template | |
| MDCG 2020-8 | C | Post-market clinical follow-up (PMCF) Evaluation Report Template | |
| MDCG 2020-5 | C | Clinical Evaluation - Equivalence | |
| ASTM D999:2015 | C | Test Methods for Vibration Testing of Shipping Containers | |
| ASTM D4728:2017 | C | Standard Test Method for Random Vibration Testing of Shipping Containers | |
| ASTM D5276:2017 | C | Standard Test Method for Drop Test of Loaded Containers by Free Fall | |
| ASTM D4169:2016 | C | Standard Practice for Performance Testing of Shipping Containers and Systems | |
| ASTM D4332:2014 | C | Standard Practice for Conditioning Containers, Packages or Packaging Components for Testing | |
| ASTM F2213 (2017) | C | Standard Test Method for Measurement of Magnetically Induced Torque on Medical Devices in the Magnetic Resonance Environment | |
| ETSI EN 300-328:2019 | C | Electromagnetic Compatibility and radio spectrum matters (ERM) - Wideband Transmission Systems - Data transmission equipment operating In The 2.4 GHz ISM band and using wide band modulation techniques | |
| ETSI EN 301 489-1 v2.2.3 | C | ElectroMagnetic Compatibility (EMC) standard for radio equipment and services – Part 1: Common technical requirements – Harmonised Standard for ElectroMagnetic Compatibility | |
| ETSI En 301 489-1-17 v3.2.4 | C | ElectroMagnetic Compatibility (EMC) standard for radio equipment and services - Part 17: Specific conditions for Broadband Data Transmission Systems - Harmonised Standard for ElectroMagnetic Compatibility | |
| UN DOT 38.3 | C | Safety of primary and secondary lithium cells and batteries during transport | |
| HIPAA (1996) | R | Health Insurance Probability and Accountability Act | |
| GDPR 2016/679 (2016) | C | General Data Protection Regulation | |
| RoHS recast Directive 2011/65/EU (2011) + Amd Directive 2017/2102/EU | C | Restricting the use of hazardous substances in electrical and electronic equipment | |
| WEEE Directive 2012/19/EU | C | Waste Electrical & Electronic Equipment (WEEE) Directive | |
| ISO/IEC 27001:2017 | R | Information technology - security techniques - information security management systems - requirements | |
| ISO 27799:2016 | R | Health informatics — Information security management in health using ISO/IEC 27002 | |
| FDA Guidance (2002) | R | General Principles of Software Validation; Final Guidance for Industry and FDA Staff | |
| FDA Guidance (2016) | R | Applying Human Factors and Usability Engineering to Medical Devices | |
| FDA Guidance (2005) | R | Guidance for the Content of Pre-Market Submissions for Software Contained in Medical Devices | |
| FDA Guidance (2017) | R | Deciding When to Submit a 510(k) for a Change to an Existing Device | |
| FDA Guidance (2016) | R | Use of International Standard ISO 10993-1, “Biological evaluation of medical devices - Part 1: Evaluation and testing within a risk management process” | |
| FDA Guidance (2019) | R | Safety and Performance Based Pathway | |
| FDA Guidance (2013) | R | Radio Frequency Wireless Technology in Medical Devices | |
| FDA Guidance (2018) | R | Content of Premarket Submissions for Management of Cybersecurity in Medical Devices (DRAFT) | |
| FDA Guidance (2016) | R | Postmarket Management of Cybersecurity in Medical Devices | |
| FDA Guidance (2016) | R | Unique Device Identification System: Form and Content of the Unique Device Identifier (UDI) | |
| FDA Guidance (2017) | R | Reprocessing Medical Devices in Health Care Settings: Validation Methods and Labeling | |
| AAMI TIR 12 (2010) | C | Designing, testing, and labeling reusable medical devices for reprocessing in health care facilities: A guide for medical device manufacturers | |
| AAMI TIR 30 (2011 /(R)2016) | C | A compendium of processes, materials, test methods, and acceptance criteria for cleaning reusable medical devices | |
| AAMI TIR 69 (2017) | C | Risk Management of Radio-frequency Wireless Coexistence for Medical Devices and Systems | |
| FCC part 15 Subpart C | C | FCC: Rules and regulations for Title 47 (Telecommunications) | |
| EN 301 489-17 v3.2.4 (2020-09) | C | EMC Testing | |
| FCC Part 15 Subpart B | C | EMC Testing | |
| NBOG (2014) | R | Consensus paper for the Interpretation and Application of Annexes Z in EN ISO 14971 2012 | |
| NBOG BPG 2014-3 (2014) | R | Guidance for manufacturers and Notified Bodies on reporting of Design Changes and Changes of the Quality System |